Psychol Med (2001) 31: 3-14The molecular biology of Huntington´s disease.
http://nickelsgen677s09.weebly.com/uploads/1/7/2/1/1721478/4708686.jpg?472x328
{kind=link}
LW Ho, J Carmichael, J Swartz, A Wyttenbach, J Rankin, DC Rubinsztein
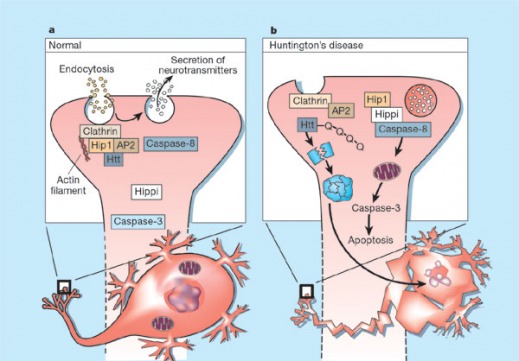
BACKGROUND: Huntington's disease (HD) is a fatal neurodegenerative disorder with an autosomal dominant mode of inheritance. It leads to progressive dementia, psychiatric symptoms and an incapacitating choreiform movement disorder, culminating in premature death. HD is caused by an increased CAG repeat number in a gene coding for a protein with unknown function, called huntingtin. The trinucleotide CAG codes for the amino acid glutamine and the expanded CAG repeats are translated into a series of uninterrupted glutamine residues (a polyglutamine tract). METHODS: This review describes the epidemiology, clinical symptomatology, neuropathological features and genetics of HD. The main aim is to examine important findings from animal and cellular models and evaluate how they have enriched our understanding of the pathogenesis of HD and other diseases caused by expanded polyglutamine tracts. RESULTS: Selective death of striatal and cortical neurons occurs. It is likely that the HD mutation confers a deleterious gain of function on the protein. Neuronal intranuclear inclusions containing huntingtin and ubiquitin develop in patients and transgenic mouse models of HD. Other proposed mechanisms contributing to neuropathology include excitotoxicity, oxidative stress, impaired energy metabolism, abnormal protein interactions and apoptosis. CONCLUSIONS: Although many interesting findings have accumulated from studies of HD and other polyglutamine diseases, there remain many unresolved issues pertaining to the exact roles of intranuclear inclusions and protein aggregates, the mechanisms of selective neuronal death and delayed onset of illness. Further knowledge in these areas will inspire the development of novel therapeutic strategies.
http://hdlighthouse.org/research/brain/updates/1229pathogenesis.php
Inga kommentarer:
Skicka en kommentar